レケンビ
※ご使用にあたって、また、「警告・禁忌を含む注意事項等情報」等については電子添文をご参照ください
試験概要
国際共同第III相プラセボ対照比較試験BAN2401-G000-301試験 Core Study(301試験Core Study)(国際共同試験、海外データを含む)7,8)
8)[承認時評価資料]国際共同臨床第III相試験(301試験)[LEQ-0009]
7)van Dyck CH, et al.: N Engl J Med. 2023; 388(1): 9-21. [LEQ -0002]
[利益相反:本試験はエーザイ(株)の支援によって行われた。著者にエーザイ(株)の社員が含まれる。]
目的
早期アルツハイマー病(早期AD)患者 ※1を対象に、Clinical Dementia Rating-Sum of Boxes(CDR-SB)を指標として、レケンビ10㎎/kg隔週投与のプラセボに対する優越性を検証し、その有効性を評価するとともに、安全性を評価した。
※1:アルツハイマー病による軽度認知障害(MCI due to AD)の可能性が中等度である患者または軽度アルツハイマー型認知症患者
試験デザイン
国際共同、多施設、無作為化、二重盲検、プラセボ対照、並行群間比較
対象
早期AD患者 ※1 1795例(日本人患者152例)
主な選択基準
-
MCI due to ADの可能性が中等度
- NIA-AA※2によるMCI due to ADの中核となる臨床基準を満たし、その可能性が中等度に区分される。
-スクリーニング期およびベースライン期のCDRスコアが0.5かつCDRの記憶スコアが0.5以上。
-主観的記憶障害が潜行性に発症し、スクリーニング開始前1年の間に緩徐に進行しているとの自覚症状を有する。ただし、情報提供者によってその症状が確認される必要がある。
-
軽度アルツハイマー型認知症
-NIA-AAによるアルツハイマー型認知症(臨床的確診)の中核となる臨床基準を満たす。
-スクリーニング期およびベースライン期のCDRスコアが0.5~1.0、かつCDRの記憶スコアが0.5以上。
-
Wechsler Memory Scale-IV Logical Memory(subscale)IIの点数が年齢調整済み平均値を少なくとも1標準偏差下回り(50~64歳:15以下、65~69歳:12以下、70~74歳:11以下、75~79歳:9以下、80~90歳:7以下)、エピソード記憶障害が客観的に示される
-
スクリーニング期およびベースライン期のMMSEスコアが22以上30以下
-
性別は不問
-
年齢50~90歳
-
アミロイドPETまたはCSF検査でアミロイドβ病理を示唆する所見が認められる
※2 National Institute of Aging-Alzheimer’s Association:米国国立老化研究所-アルツハイマー病協会
主な除外基準
- 原疾患のAD以外の要因が認知機能障害に影響を及ぼし得る状態にある
-
治験実施に支障をきたすおそれがある精神疾患または精神症状(例:幻覚、大うつ病または妄想)を有する
-
スクリーニング期のGeriatric Depression Scale(GDS)スコアが8以上
-
スクリーニング期の脳MRI検査においてAD以外の認知症を示唆する臨床的に意義のある病巣が認められる
-
スクリーニング期の脳MRI検査において、以下に示すその他の臨床的意義のある所見が認められる:
脳微小出血(最大径10mm以下)5ヵ所以上、最大径10mm超の脳出血1ヵ所、脳表ヘモジデリン沈着症、血管原性脳浮腫、脳挫傷、脳軟化、動脈瘤、血管奇形、感染病巣、多発性ラクナ梗塞または大血管支配領域の脳卒中、重度の小血管疾患または白質疾患または占拠性病変または脳腫瘍(ただし、髄膜腫またはくも膜嚢胞と診断される病変で、最大径が1cm未満であれば除外する必要はないこととした) -
レカネマブの投与歴を有する
| 5. 効能又は効果に関連する注意(一部抜粋) 5.2. 承認を受けた診断方法、例えばアミロイドPET、脳脊髄液(CSF)検査、又は同等の診断法によりアミロイドβ病理を示唆する所見が確認され、アルツハイマー病と診断された患者のみに本剤を使用すること。 |
方法
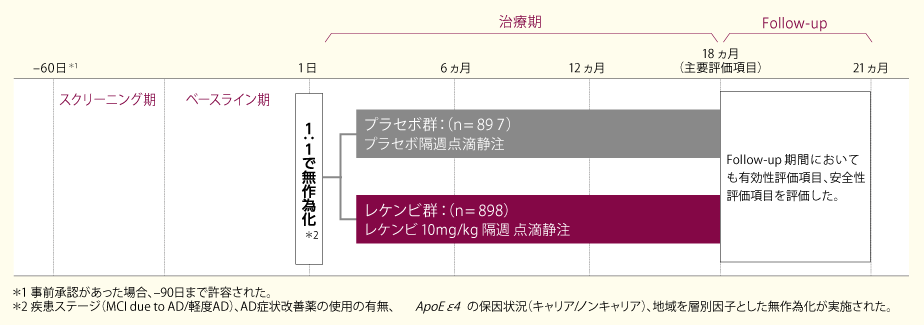
スクリーニング期に選定された1795例を無作為化し(プラセボ群897例、レケンビ群898例)、隔週にて18ヵ月間投与した。
プラセボ(プラセボ群)またはレケンビ10mg/kg(レケンビ群)を、それぞれ隔週ごとに60分間かけて点滴静注した。
評価項目
有効性※3
主要評価項目(検証的解析結果)
投与18ヵ月後におけるCDR-SBのベースラインからの変化量
重要な副次評価項目
投与18ヵ月後におけるADCS MCI-ADL、ADCOMS、ADAS-Cog14、アミロイドPETセンチロイドスケールを指標とした脳内アミロイドβ蓄積量のベースラインからの変化量
探索的評価項目
治療期の18ヵ月間におけるCDRスコアの悪化までの期間、投与18ヵ月後におけるQOL-ADおよびZBIのベースラインからの変化量など
バイオマーカー評価項目
投与後3、6、12ヵ月におけるアミロイドPETセンチロイドスケールを指標とした脳内Aβ蓄積量のベースラインからの変化量、投与12ヵ月後および18ヵ月後における脳脊髄液(CSF)中のAD関連バイオマーカー(Aβ42 、Aβ40 、ニューログラニン、NfL ※4 、t-tau、p-tau181 ※5 ) など
安全性
有害事象、臨床検査、バイタルサイン、心電図、脳MRI、コロンビア自殺評価スケール(C-SSRS ※6 )
※4:neurofilament light chain(ニューロフィラメント軽鎖)
※5:human tau protein phosphorylated at threonine in position 181(リン酸化タウ181)
※6:Columbia-Suicide Severity Rating Scale
解析計画
投与18ヵ月後におけるCDR-SBのベースラインからの変化量の主解析では、FAS(full analysis set)+ ※7を対象に、経時的反復測定データに対する混合効果モデル(MMRM)※8 を用いてレケンビ群とプラセボ群を比較した。
MMRMには、CDR-SBのベースライン値を共変量として、投与群、評価時点、無作為化時の層別因子(疾患ステージ[MCI due to ADまたは軽度アルツハイマー型認知症]、AD症状改善薬のベースライン時の併用有無[アセチルコリンエステラーゼ 阻害薬<AChEI>および/またはメマンチン]、ApoE ε4 保因状況[キャリアまたはノンキャリア]、地域[北米、オーストラリアを含む欧州、中国を除くアジア太平洋地域])、CDR-SBのベースライン値と評価時点との交互作用、並びに投与群と評価時点との交互作用項を固定効果として含めた。欠測値の補完は行わなかった。帰無仮説は、投与18ヵ月後におけるCDR-SBのベースラインからの平均変化量にレケンビ群とプラセボ群との間で差がないとし、有意水準を両側0.05として検証した。
重要な副次評価項目についても、上記と同様の統計手法を用いて解析した。なお、アミロイドPETセンチロイドスケールに関する重要な副次評価項目の解析にはPD解析対象集団 ※9を用いた。
※7:無作為化後に治験薬が投与され、主要評価項目の評価が可能なデータ(ベースラインおよび治験薬投与後)を有する被験者集団とした。なお、新型コロナウイルス感染症(COVID-19)パンデミック中(2020年3月1日~2020年7月31日)に6週間(42日、すなわち連続する3回の投与に該当)以上、治験薬の投与が停止された実施医療機関で、当該停止期間終了よりも前に無作為化された被験者をFAS+から除外した集団をFASとした。
※8:変量効果と固定効果を含む線形混合効果モデルに基づき不完全な経時測定データを解析するために利用される統計モデルであり、観測データや対象者のベースライン情報等から欠測データを予測することで調整済み平均値を算出する。
※9:治験薬が投与され、1つ以上の薬力学パラメータを算出することが可能なデータ(ベースラインおよび治験薬投与後)を有する被験者集団と
した。